- +1
專訪許錦波:預測蛋白質(zhì)結(jié)構(gòu)二十余載,這條路如何從冷清到熱鬧
DNA儲存著我們的遺傳信息,然而在細胞中真正執(zhí)行功能的是蛋白質(zhì)。每個蛋白質(zhì)的氨基酸鏈扭曲、折疊、纏繞成復雜的結(jié)構(gòu),“看清”它們的結(jié)構(gòu)對理解其功能至關(guān)重要。但想要破解這種結(jié)構(gòu)通常需要花很長的時間,有些甚至難以完成。
“用機器學習去研究蛋白質(zhì)結(jié)構(gòu)預測,在這個領(lǐng)域?qū)儆谏贁?shù)派。一直到2016年,甚至到2018年,這個領(lǐng)域大部分人都還在試圖用能量優(yōu)化,而不是機器學習或者深度學習去研究這個問題。”美國芝加哥豐田計算技術(shù)研究所教授、北京大學客座教授許錦波在接受澎湃新聞(www.6773257.com)記者專訪時如是表示。
許錦波被業(yè)界譽為“AI預測蛋白質(zhì)結(jié)構(gòu)全球第一人”。早在2016年,他開發(fā)的RaptorX-Contact方法,首次證明了深度學習方法預測蛋白質(zhì)結(jié)構(gòu)的可行性,讓始終在“門口”徘徊的蛋白質(zhì)結(jié)構(gòu)預測終于邁出實質(zhì)性的一步,也自此掀起了AI蛋白質(zhì)結(jié)構(gòu)預測的熱潮。

美國芝加哥豐田計算技術(shù)研究所教授、北京大學客座教授許錦波。
現(xiàn)年48歲的許錦波從小就是一名不折不扣的“學霸”。1990,16歲的許錦波在全國高中數(shù)學聯(lián)賽中獲江西賽區(qū)第一名,這也是當時江西臨川縣首次獲得該類獎項殊榮。1991年,因為在數(shù)學競賽中的優(yōu)異成績,他從臨川一中被保送至中國科學技術(shù)大學計算機系,1999年獲得中國科學院計算所碩士學位。2003年,許錦波獲加拿大滑鐵盧大學博士學位,之后任該校研究助理教授、麻省理工學院博士后研究員。
2001年,尚在攻讀博士學位的許錦波開始接觸計算生物學,彼時的導師提議,“有一個很難的問題,就是研究蛋白質(zhì)折疊,想不想做?”在此后的二十余年時間里,許錦波研究的重要課題之一就是開發(fā)和優(yōu)化軟件,去無限縮小蛋白質(zhì)結(jié)構(gòu)預測結(jié)果和真實構(gòu)型之間的差距。
近日,在未來論壇主辦的2022《理解未來》科學講座01期“AI+蛋白質(zhì)結(jié)構(gòu)和功能預測”上,許錦波也首先談到,其實蛋白質(zhì)結(jié)構(gòu)預測這個問題已經(jīng)研究了幾十年,過去這個領(lǐng)域一直比較冷清,特別是在2006年到2016年這10年間,“當時大家都覺得這個問題是沒辦法做出來的,所以很多人都離開這個領(lǐng)域去做其他的問題了。”
這樣的冷清已經(jīng)是過去式。在最近的幾年時間里,這一領(lǐng)域陸續(xù)獲得突破性的進展。2020 年,人工智能預測蛋白質(zhì)結(jié)構(gòu)也被國際頂級學術(shù)期刊《科學》雜志評為十大科學突破之一。“現(xiàn)在人工智能預測蛋白質(zhì)結(jié)構(gòu)受到的關(guān)注,遠遠超過了過去幾十年來的關(guān)注。”許錦波表示。
然而,在冷清的路上走慣了的許錦波,對眼下的熱鬧并沒有表現(xiàn)出太多的興奮。談及這兩年陸續(xù)成立的人工智能應(yīng)用于生命科學領(lǐng)域的公司,他坦言,“我對產(chǎn)業(yè)的了解不是很多,也就最近幾個月開始接觸一些產(chǎn)業(yè)界的認識和做投資的人。”當然,許錦波認為,對于“AI For science”的產(chǎn)業(yè)化而言,當下的確處于一個比較好的時候。
但許錦波強調(diào),就人工智能預測蛋白質(zhì)結(jié)構(gòu)而言,重復實現(xiàn)明星公司DeepMind的AlphaFold2不應(yīng)該成為其他團隊的目標,“這種改進只是一個漸進式的改進,并不是一個非常大的突破,這個領(lǐng)域仍然有一系列問題真正需要我們?nèi)ソ鉀Q。”對于人工智能在藥物研發(fā)等生命領(lǐng)域的應(yīng)用,他則表示,“希望能夠做出一些真正有用的東西出來。”
始于半個世紀前的猜測
蛋白質(zhì)結(jié)構(gòu)預測,始于科學家們的一種設(shè)想,是否無需實驗就能獲取蛋白質(zhì)的三維結(jié)構(gòu)?
在蛋白質(zhì)結(jié)構(gòu)解析的幾十年歷史中,結(jié)構(gòu)生物學家們用X射線晶體學、核磁共振波譜學(NMR)、冷凍電鏡(Cryo-SEM)技術(shù)解析了很多蛋白的結(jié)構(gòu),并以此更好地推進疾病機理、藥物研發(fā)等工作。
然而,這些手段被視作勞心勞力又價格高昂。截至目前,約有10萬個蛋白質(zhì)的結(jié)構(gòu)已經(jīng)用實驗方法得到了解析,但這在已經(jīng)測序的數(shù)10億計的蛋白質(zhì)中只占了很小一部分。
作為學計算機出身的一名科學家,許錦波對他研究了近20年的蛋白質(zhì)如此理解:蛋白質(zhì)是由很多氨基酸通過化學鍵串聯(lián)在一起,如果把每個氨基酸看成一個珠子的話,那么就有20種不同顏色的珠子,這些珠子串在一起形成蛋白質(zhì)的氨基酸系列,每一個不同的顏色用一個字母表示,所以蛋白質(zhì)氨基酸序列可以看成是1個由20個字母組成的字符串。每個氨基酸又是由幾十個原子組成的,所以整個蛋白質(zhì)是由成千上萬個原子構(gòu)成的,這些原子在細胞里面有相互作用力,最后形成一個穩(wěn)定的構(gòu)型。
“我們可以用不同的軟件去把這些結(jié)構(gòu)給顯示出來,但是在利用這些軟件去顯示蛋白質(zhì)構(gòu)型的時候,我們需要知道這些原子在三維空間中的位置,需要知道它們的三維坐標,怎么樣才能知道這些三維坐標?”許錦波提到,在過去很多年里,科學家發(fā)展了三種主要的實驗技術(shù),去測定這些原子的三維坐標。
除了上述提到的三種實驗室技術(shù)之外,科學家們也在研究,計算方法的推導是否可行?
許錦波對澎湃新聞記者表示,美國生物化學家、1972年諾貝爾化學獎得主克里斯蒂安·安芬森(Christian Boehmer Anfinsen)通過實驗提出了他自己的猜想,“這位實驗學家的猜測基本是對的,他自己做了一些列實驗支持了這個理論。”
安芬森的工作大部分圍繞蛋白質(zhì)的結(jié)構(gòu)與功能之間的關(guān)聯(lián)性。1961年,他研究了核糖核酸酶可以在變性后重新進行折疊,恢復到原來的空間結(jié)構(gòu),同時保留酵素的活性。安芬森因此認為,所有造成最終構(gòu)象所需的蛋白質(zhì)信息都被編碼于其氨基酸序列上,即蛋白質(zhì)一級排序決定三維結(jié)構(gòu)。
上述即被稱為安芬森法則,這也是蛋白質(zhì)結(jié)構(gòu)預測的基石。

美國生物化學家、1972年諾貝爾化學獎得主克里斯蒂安·安芬森。
然而,在隨后的50多年時間里,科學家們使用了各種各種的方法,都無法精確計算蛋白質(zhì)的三維結(jié)構(gòu)。“在安芬森這個假設(shè)和理論基礎(chǔ)之下,科學家們?nèi)プ龅鞍踪|(zhì)折疊預測,都是從能量優(yōu)化的角度去做。”許錦波解釋,大家普遍認為,蛋白質(zhì)是折疊到最小能量狀態(tài),這也意味著,從理論上來說,如果能更好地優(yōu)化這個能量函數(shù),就能夠找到蛋白質(zhì)的最小能量狀態(tài)。
但這一思路有著天然缺陷。“第一,一個蛋白質(zhì)是一個非常大的體系,由成千上萬個原子組成,對應(yīng)一個非常巨大的搜索空間,構(gòu)型是千變?nèi)f化的。”許錦波繼續(xù)提出第二個困難之處,“雖然說大家普遍接受蛋白質(zhì)折疊到最小能量狀態(tài),但能量函數(shù)到底是什么樣的?我們本身就對能量函數(shù)的理解還不是特別好。”
許錦波在博士階段最初也是使用傳統(tǒng)的優(yōu)化算法去研究這一問題。2001年,他接下了導師向他拋出的這一課題,第二年即取得了不錯的成果,在2002年全球蛋白質(zhì)結(jié)構(gòu)預測比賽CAFASP(用于全自動高通量蛋白質(zhì)結(jié)構(gòu)預測的評比)中,奪得冠軍。
回憶當時的成績,許錦波略顯輕描淡寫,“雖然排名最好,但是意義并沒有那么大,并沒有改變這個問題的現(xiàn)狀,只是結(jié)果比別人好一點點而已。”在這一思路下繼續(xù)了一年多之后,他意識到,傳統(tǒng)的優(yōu)化算法可能不是一個很好的路徑。
2006年,許錦波開始轉(zhuǎn)向機器學習,彼時已組建獨立實驗室的他認為,應(yīng)該改變策略。“我們用機器學習做的比傳統(tǒng)的方法好一點,在蛋白質(zhì)結(jié)構(gòu)預測比賽中,也取得了很好的成績,比別的組要好一點,但也并沒有特別大的改變。”
這條路徑一走就又是8年,應(yīng)該也是許錦波科研道路上最冷清的8年,很多人陸續(xù)轉(zhuǎn)行,領(lǐng)域幾無關(guān)注。
人工智能為什么可以成功
2014年,許錦波開始第二次轉(zhuǎn)換途徑。
“2012年,深度學習開始在圖像識別中做到了很好的結(jié)果,所以我們在2014年開始嘗試用深度學習去研究這個問題。”真正將AI納入到許錦波預測蛋白質(zhì)結(jié)構(gòu)的工具箱中,始于這一年。彼時,同領(lǐng)域中只有極少數(shù)人關(guān)注到這一新的工具。
“新方法不是去做能量最優(yōu)化,而是預測原子之間的相互作用關(guān)系。”
許錦波進一步解釋道,假設(shè)已有一個氨基酸序列,那么把和這一蛋白質(zhì)同源(同一個家族)的那些蛋白質(zhì)都找出來,然后把所有這些同一個家族的蛋白質(zhì)的氨基酸序列都比對在一起。“多序列對比下,我們用矩陣去表示蛋白質(zhì)里面氨基酸之間相互作用關(guān)系,然后根據(jù)相互作用關(guān)系矩陣,就可以把蛋白質(zhì)的原子的坐標預測出來,這是這種新方法的總體思路。”
當然,在總體思路框架下可以有不同的實現(xiàn)方法,“但新方法的關(guān)鍵點在于,我們能不能準確地推斷出蛋白質(zhì)里面原子之間或者氨基酸之間的相互作用關(guān)系,這一步是非常關(guān)鍵的。”
許錦波談到,為了預測原子之間的相互作用關(guān)系,科學家們探索的最早方法是協(xié)同進化全局統(tǒng)計方法(global statistical method for co-evolution analysis)。然而,這種方法只對極少比例蛋白質(zhì)有效,而往往這些蛋白質(zhì)家族里某些蛋白的三維結(jié)構(gòu)已經(jīng)被實驗技術(shù)測出來了,這也意味著用這種方法預測的意義并不太大。
他認為,真正對大量的蛋白質(zhì)結(jié)構(gòu)預測其作用的轉(zhuǎn)折之年是2016年。在轉(zhuǎn)向深度學習2年之際,許錦波開始用深度學習預測蛋白質(zhì)的三維結(jié)構(gòu)。而在此前的2年時間里,其團隊以更為簡單的問題入手,嘗試預測蛋白質(zhì)的二級結(jié)構(gòu),即肽鏈主鏈骨架原子的空間位置排布,不涉及氨基酸殘基側(cè)鏈。
“對于這么一個簡單的問題能夠做得好,我們認為對于更難的問題,也就是預測蛋白質(zhì)的三維結(jié)構(gòu)應(yīng)該會有效果。”許錦波提到一個細節(jié),在2015年其就組織學生去解決三維結(jié)構(gòu)的問題,然而并沒有實現(xiàn),“他們不太理解我的想法,因為那個時候在這個領(lǐng)域沒有人用深度卷積網(wǎng)絡(luò)去解決這個問題。”
2016年,騰出一些時間的許錦波開始自己寫代碼去實現(xiàn)自己的算法,“大概在那年暑假的時候就得到了非常好的結(jié)果,發(fā)現(xiàn)一下子能做得比以前的方法好非常多,2016年秋天,我把結(jié)果寫成一篇論文發(fā)布在了網(wǎng)上。”發(fā)布后的第一個月,即在領(lǐng)域內(nèi)引起了一波關(guān)注高潮。
許錦波發(fā)布的正是他開發(fā)的第一代人工智能方法RaptorX。該方法基本的原理是,通過深度卷積殘差網(wǎng)絡(luò)(ResNet),對蛋白質(zhì)的序列進行卷積變換,從中抽取出有效信息,同時也對蛋白質(zhì)殘基之間相互作用關(guān)系進行卷積變換。通過這兩者不同的卷積變換,可以非常準確地預測蛋白質(zhì)氨基酸之間的相互作用關(guān)系。“然后基于這個相互作用關(guān)系,我們可以把它的三維結(jié)構(gòu)重構(gòu)出來。”
在2016年全球蛋白質(zhì)結(jié)構(gòu)預測比賽(CASP12)中,這一尚未完善好的方法即嶄露頭角,“當時已經(jīng)做得非常好,做的比其他傳統(tǒng)方法都要好。”

2017年1月,許錦波將前期成果正式發(fā)表于《PLOS Computational Biology 》,題為“Accurate De Novo Prediction of Protein Contact Map by Ultra-Deep Learning Model”。在這篇論文中,研究團隊展示了通過使用深度殘差卷積網(wǎng)絡(luò),可以大幅度提高蛋白質(zhì)預測的精度,并且這種學習方法也很容易推廣到不同類型的蛋白質(zhì)層面,比如一些膜蛋白及蛋白復合物等的結(jié)構(gòu)。
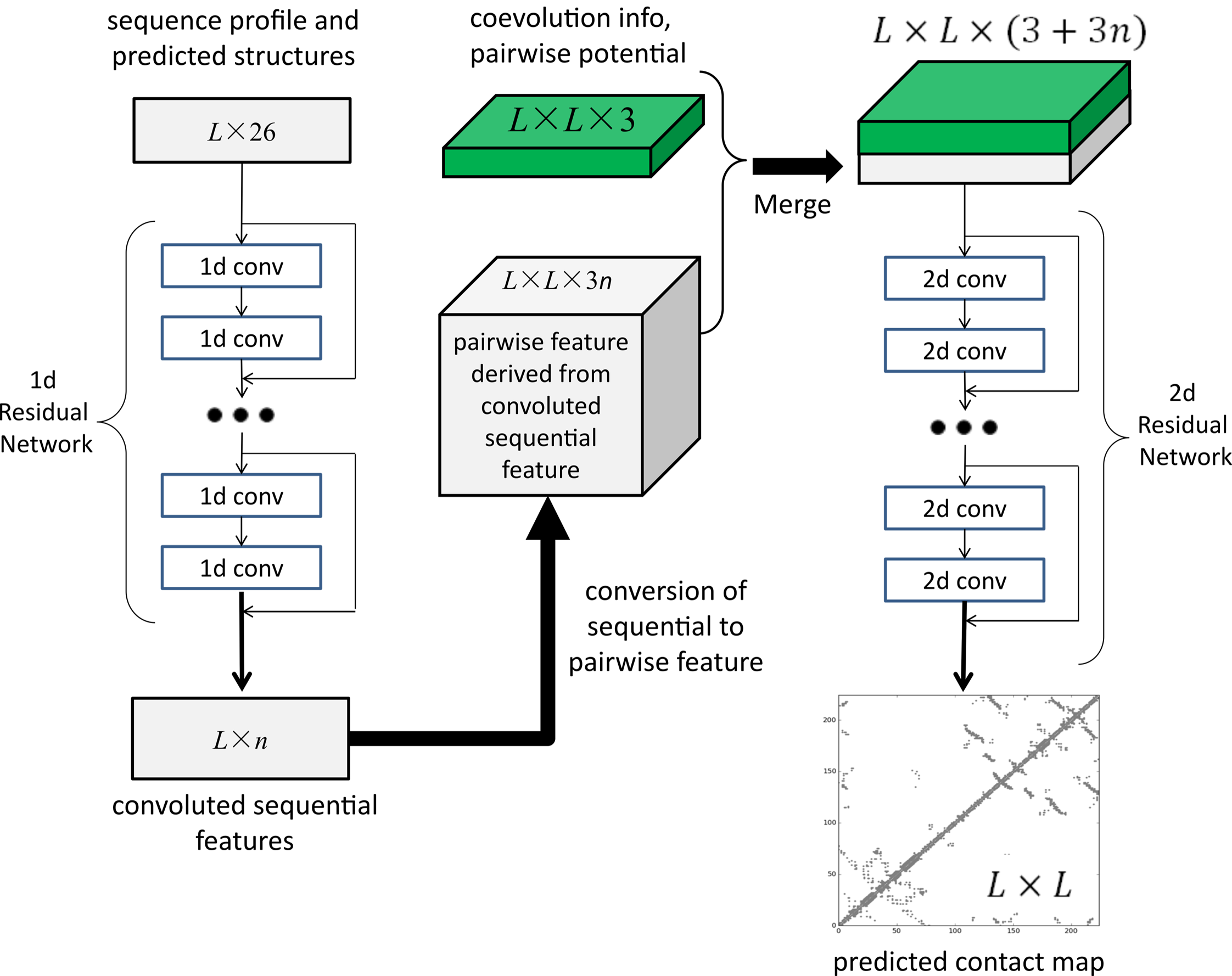
至今這仍是許錦波最滿意的一篇論文。“我們論文出來之后,其實把問題定義得很清楚了。從AI的角度來說,就是告訴大家這個問題的輸入是什么,輸出是什么,你只要把AI算法做好就行了。至于你用什么AI算法,無非更多的是工程上和計算資源上的問題。”
他還向澎湃新聞記者回憶了一段小插曲,研究團隊實際上最開始將論文投到了《自然》(Nature)的一本子刊,然而編輯并不太相信他們的結(jié)果。“因為這個問題研究很多年了,一直沒有什么進展,他不認為我們能做得這么好,另外一本期刊的一個評委都不認為我們的結(jié)果是可靠的。”
令許錦波欣慰的是,無論是學術(shù)界還是產(chǎn)業(yè)界,都在論文發(fā)表之后對該研究給予了廣泛的關(guān)注。他感受到,總體而言,學計算機出身的人更容易接受他們的結(jié)果,而學生物化學或者生物物理的人,因為此前就不習慣于使用類似的方法,并不太容易接受這項結(jié)果。
值得一提的是,在蛋白質(zhì)結(jié)構(gòu)預測領(lǐng)域過去近30年的時間里,該領(lǐng)域的發(fā)展大致可以分三個階段。第一個階段,也就是長達20多年的時間里,在傳統(tǒng)方法之下該領(lǐng)域進展非常緩慢;第二個階段,也就是通過使用許錦波等人開發(fā)的第一代人工智能方法RaptorX,難度較大的蛋白質(zhì)結(jié)構(gòu)的預測精度已被大幅提升;而在第三個階段,則是目前為止全球表現(xiàn)最好的蛋白質(zhì)結(jié)構(gòu)預測工具,也就是DeepMind在2020年推出的AlphaFold2。“通過使用注意力機制網(wǎng)絡(luò),又可以大幅度提高蛋白質(zhì)結(jié)構(gòu)預測的精度。”
在許錦波看來,DeepMind在2017年、2018年之際,實際上在重新實現(xiàn)他的算法,“當然他們工程上做得比我們好一些。”而對于DeepMind在AlphaFold2中使用的注意力機制網(wǎng)絡(luò),其最早被應(yīng)用于自然語言處理中。
“計算生物學領(lǐng)域的人知道的并不是很多,最早將這一網(wǎng)絡(luò)真正用到這個領(lǐng)域的是Facebook,他們沒有用來做蛋白質(zhì)結(jié)構(gòu)預測,而是用來對蛋白質(zhì)序列進行建模。”許錦波提到,即使后來計算生物學領(lǐng)域的人注意到了基于注意力機制的網(wǎng)絡(luò),然而該網(wǎng)絡(luò)需要太多的計算資源,“學術(shù)界沒有人有這么多資源去做這件事情。”
許錦波坦言,其團隊在2020年曾經(jīng)考慮如何簡化基于注意力機制的網(wǎng)絡(luò),“希望使它能夠在我們的計算資源上跑起來,這是我當時做的事情,因為我們沒有幾百塊GPU(顯卡上的芯片)。”相比之下,背靠谷歌的DeepMind完全沒有這方面的“資源窘境”,可以用很多GPU卡訓練他們的模型。
許錦波認為,從思想創(chuàng)新而言,AlphaFold2邁的這一步并不沒有讓人感到非常吃驚的。“真正吃驚的是他們能夠一下子調(diào)動30個人去做這個事情,能夠把它實現(xiàn)得非常好,我覺得這是他們的長處。”
總體而言,人工智能對蛋白質(zhì)結(jié)構(gòu)預測領(lǐng)域起到了非常大的推動作用,而過去這么多年里,為何又只有深度學習能夠做到?
許錦波分享了他個人的理解,首要的前提是,深度學習是基于現(xiàn)有的理論基礎(chǔ),特別是進化論。“第一,雖然我們沒有它們的構(gòu)型,但是我們知道,同一個家族的蛋白質(zhì)結(jié)構(gòu)應(yīng)該是很相似的。第二,同一個蛋白里面空間中相鄰的氨基酸互相影響、共同進化,這點也非常重要。”
除理論基礎(chǔ)外,許錦波認為對于訓練深度學習算法而言,數(shù)據(jù)當然必不可少。“現(xiàn)在我們有了大量的蛋白質(zhì)序列數(shù)據(jù),可以依據(jù)同一個家族里面蛋白質(zhì)的進化關(guān)系去推斷原子在空間中的距離,這是非常重要的。另外一個很重要的數(shù)據(jù)源是我們也有了一些蛋白質(zhì)結(jié)構(gòu)數(shù)據(jù),雖然說沒有那么多,但現(xiàn)在我們至少有一些,那么通過指導深度學習模型去學習氨基酸共進化與原子間中距離的關(guān)系。”
比重復實現(xiàn)AlphaFold2更重要的事情
尤其在AlphaFold2出現(xiàn)之后,人工智能預測蛋白質(zhì)結(jié)構(gòu)這一領(lǐng)域受到了空前的關(guān)注,終于“熱鬧”了起來。
許錦波總結(jié)認為,人工智能的確顛覆了蛋白質(zhì)結(jié)構(gòu)預測,而這會帶來非常大的改變,尤其對分子生物學科來說,“我想這個結(jié)果現(xiàn)在已經(jīng)改變了很多分子生物學家的研究范式,以前的分子生物學家基本都基于蛋白質(zhì)的氨基酸序列去分析蛋白質(zhì)的功能,現(xiàn)在很多人都開始使用預測的結(jié)構(gòu)去做研究、去分析蛋白質(zhì)的功能,所以這是一個非常大的研究范式的改變。”
但現(xiàn)在還遠遠沒有到達終點,將來又如何繼續(xù)推進人工智能在結(jié)構(gòu)生物學甚至更廣泛的生物學中的應(yīng)用?
許錦波談道,有很多團隊在致力于重復實現(xiàn)AlphaFold2,“當然這是一條必經(jīng)之路,但這種改進只是一種漸進式的改進,即使我們能夠做的好一點點,其實也不是一個非常大的突破。”他同時提醒,如果很多團隊或者初創(chuàng)公司一窩蜂去做這件事情,“我覺得有點浪費資源。”
在他看來,那些當下解決得還不夠好的問題,需要去真正地投入更多的精力。
例如,我們能不能對一個孤兒蛋白進行非常準確預測?能不能預測蛋白質(zhì)的折疊過程,而不僅僅是最后構(gòu)型?能不能準確預測蛋白質(zhì)復合物或者一個多域蛋白的結(jié)構(gòu)?能不能預測蛋白質(zhì)和多肽、DNA或者RNA的相互作用?能不能預測單點或多點突變對一個蛋白質(zhì)結(jié)構(gòu)和功能的影響?
他對澎湃新聞記者進一步表示,我們對蛋白質(zhì)結(jié)構(gòu)預測的要求取決于我們的目標。如果目標只是想知道這個蛋白質(zhì)最終的三維形狀,對于大部分蛋白質(zhì)來說其實已經(jīng)做到了這一點。“然而現(xiàn)在我們能做的,就是可以把單個蛋白的結(jié)構(gòu)預測得很好。但是對于蛋白質(zhì)復合物等更加復雜的情況,人工智能的方法確實能做得比以前好很多,但是還沒有達到讓人非常滿意的狀態(tài),這個方向還需要花更多的時間去研究。”
許錦波同時拋出一個更值得思考的問題,“現(xiàn)在所有的成功方法其實都有點cheating。”這也是一個從原理上即存在的問題。
不難理解,如此說的原因在于,目前的方法需要使用大量的蛋白質(zhì)同源信息,“能夠找到越多的同源蛋白,這種預測效果越好。如果沒有這部分的信息,現(xiàn)在所有的方法都沒有效果。”許錦波說,在細胞里面,也就是自然界的蛋白質(zhì)在折疊的時候,“它并不知道同家族到底有哪些蛋白質(zhì),它自己能夠折疊出來,它不需要知道有多少‘兄弟姐妹’。”
值得一提的是,許錦波已經(jīng)回國,并決定將重心轉(zhuǎn)移到國內(nèi)。“創(chuàng)新驅(qū)動發(fā)展戰(zhàn)略是我們國家綜合國力發(fā)展的有力保障,”許錦波對澎湃新聞記者表示,“我希望做一些真正原創(chuàng)且能落地的東西出來,推動科研與產(chǎn)業(yè)化的融合發(fā)展。”
談到“AI+生命科學”的產(chǎn)業(yè)應(yīng)用價值,許錦波表示,目前“AI for Science”的產(chǎn)業(yè)化環(huán)境很好,特別是“AI for BioTech”。“國家在‘AI for BioTech’領(lǐng)域非常重視,投資機構(gòu)也非常支持硬科技領(lǐng)域的早期、長期投資。”而從產(chǎn)業(yè)角度來講,他認為,由于AI在生物制藥領(lǐng)域為各個環(huán)節(jié)賦能,幫助行業(yè)提升了效率與準確度,因此AI在該領(lǐng)域的產(chǎn)業(yè)化也具有很好的前景。
值得關(guān)注的是,今年1月,許錦波在北京創(chuàng)立北京分子之心科技有限公司(下稱“分子之心”)。就在4月,該公司宣布已完成數(shù)千萬美元天使輪融資,由紅杉中國領(lǐng)投,百度風投、生命園創(chuàng)投基金、NeuX Capital芯航資本 、未來啟創(chuàng)基金等跟投。分子之心稱,該輪融資將用于進一步擴大團隊、AI蛋白質(zhì)平臺的持續(xù)進化,以及科研成果的產(chǎn)品化轉(zhuǎn)化。
他對澎湃新聞記者表示,公司目前僅有一個很小的團隊在繼續(xù)研究蛋白質(zhì)結(jié)構(gòu)預測的問題,“我們更主要的目標在于,能不能做各種蛋白質(zhì)的優(yōu)化和設(shè)計。比如可以把一個抗體優(yōu)化得更好,使得它能夠跟抗原結(jié)合更好;或者說能不能設(shè)計一個自然界不存在的蛋白,用它來做藥或用于其他目的;或者能不能把某一個酶優(yōu)化得更好。這是現(xiàn)在我們公司的重點。”
其最后談到,當下多學科的融合比以往更加重要,而如何吸引更多的人加入到交叉學科,同時也吸引更多的學生進入到領(lǐng)域內(nèi),這些仍面臨一些挑戰(zhàn)。
許錦波以其自身經(jīng)歷說道,“剛進入計算生物學這個領(lǐng)域的時候,我會發(fā)現(xiàn)我跟生物學家們的溝通其實是非常困難的。只有經(jīng)過一段時間之后,談話和合作才能繼續(xù)下去,多溝通多交流,我想這是非常重要的。”
而更為關(guān)鍵的一點是,他認為評估體系應(yīng)當做出一些改變。“從我的經(jīng)歷來看,做蛋白質(zhì)結(jié)構(gòu)預測或者說做計算生物學,以前其實不太受重視。之前論文都發(fā)表不到特別高影響因子的刊物上,而影響因子又跟這個領(lǐng)域多少人在做有關(guān)系。如果你用影響因子去評估一項計算生物學的工作的話,往往這些人是比較吃虧的,也進而打壓了那些做計算生物學的學生。”
許錦波的觀點是,大家應(yīng)當以比較開放的心態(tài),容忍不同領(lǐng)域人的發(fā)展。



- 報料熱線: 021-962866
- 報料郵箱: news@thepaper.cn
互聯(lián)網(wǎng)新聞信息服務(wù)許可證:31120170006
增值電信業(yè)務(wù)經(jīng)營許可證:滬B2-2017116
? 2014-2025 上海東方報業(yè)有限公司

