- +1
蛋白質結構預測哪家強?兩大國際團隊同日在頂刊開源代碼
但“看清”蛋白的結構和人類的很多疾病機理、藥物研發(fā)等等息息相關。在蛋白質結構解析的幾十年歷史中,X射線晶體學、核磁共振波譜學(NMR)、冷凍電鏡(Cryo-SEM)技術紛紛發(fā)揮了巨大的貢獻,但這些技術在科學界看來,都有著勞心勞力又價格高昂的缺點。
如何簡單地通過蛋白質的氨基酸序列來預測其形狀?如何能解答這一問題,了解生命運作方式的將打開截然不同的一扇窗。這種設想提出的50多年后,谷歌旗下人工智能公司DeepMind在去年12月的國際蛋白質結構預測競賽CASP上投下重磅,他們開發(fā)的基于神經(jīng)網(wǎng)絡的新模型AlphaFold2擊敗了其他選手,在預測準確性方面達到接近人類實驗結果,讓整個結構生物學界震驚。

北京時間7月15日,DeepMind團隊在頂級學術期刊《自然》(Nature)以“加快評審文章”(Accelerated Article Preview)形式在線發(fā)表了一篇題為“Highly accurate protein structure prediction with AlphaFold”的論文,全面詳述了半年前造成轟動的這一模型,并首次對外分享開源代碼。該論文于今年5月11日提交,7月12日被接收。
DeepMind團隊向澎湃新聞(www.6773257.com)記者提供了一份聲明,公司創(chuàng)始人兼首席執(zhí)行官Demis Hassabis在聲明中表示,去年在CASP14大會上我們揭曉了一個可以將蛋白質3D結構預測精確到原子水平的全新AlphaFold系統(tǒng),此后我們承諾會分享我們的方法,并為科學共同體提供廣泛、免費的獲取途徑。
“今天我們邁出了承諾的第一步,在《自然》期刊上分享AlphaFold的開源代碼,并發(fā)表了系統(tǒng)的完整方法論,詳盡細致說明AlphaFold是如何做到精確預測蛋白質3D結構的。作為一家致力于推動科學進步的公司,我們期待看到我們的方法將為科學界啟發(fā)出什么其他新的研究方法,也期待很快能和大家分享更多我們的新進展。”Hassabis表示。

值得一提的是,就在同一天,另一頂級期刊《科學》(Science)也在線發(fā)表了另一預測蛋白質結構的研究文章,題為“Accurate prediction of protein structures and interactions using a three-track neural network”。
來自華盛頓大學、哈佛大學、德克薩斯大學西南醫(yī)學中心等團隊的研究人員開發(fā)了新的深度學習工具RoseTTAFold,其擁有媲美AlphaFold2的蛋白質結構預測超高準確度,而且更快、所需計算機處理能力更低。同樣,研究團隊也對外分享了開源代碼。該論文提交于6月7日,7月7日被接收。
清華大學生命科學學院院長、高精尖中心執(zhí)行主任王宏偉對澎湃新聞(www.6773257.com)記者表示,“高質量結構預測的源代碼開放對整個科學界尤其是結構生物學領域的促進作用必然是巨大的。”他評價道,對于DeepMind這樣一家商業(yè)公司來說,“團隊愿意向公眾分享代碼,是一個新型科研范式的突破,將整體上有利于人類更好地探索未知。”
預測蛋白質結構,接近實驗室測量
50多年前,科學家們就設想用計算機預測蛋白質結構。近年來,共同演化、接觸圖預測、深度機器學習等技術的引入,一些實驗室的算法精度有了很大程度的提高。
曾經(jīng)開發(fā)出Alphago、戰(zhàn)勝人類頂尖棋手的DeepMind團隊是其中的佼佼者,其團隊的強大和資源雄厚是一般實驗室無法企及的。2020年12月1日,他們在生物領域展現(xiàn)出實力,在兩年一度的權威蛋白質結構預測評估競賽(CASP)中用AlphaFold2擊敗其他參賽團隊。
CASP是由馬里蘭大學John Moult教授等人于1994年組織。競賽使用的是最新解決且尚未在蛋白質數(shù)據(jù)庫(PDB)中存放或公開披露的結構,結構生物學家們利用X射線晶體學、核磁共振波譜學、冷凍電鏡的方法,把這些蛋白質的結構解析出來。做蛋白質結構預測的團隊則利用計算機程序來預測它們的結構。最后由獨立的科學家團隊則把計算機預測的模型和實驗室的結構對照,分析不同計算機算法的預測結果。這是一種“雙盲”測試,長期以來一直是評價結構預測準確性的金標準。
去年的CASP14共有84個常規(guī)題目,其中有14題因為生物實驗沒給出確定結構等原因被取消或延緩,其他70個題目的單體和復合物蛋白質所含有的氨基酸個數(shù)從73到2180不等。
19個國家的215個小組參加了CASP14。DeepMind公司的AlphaFold2預測的大部分結構達到了空前的準確度,不僅與實驗方法不相上下,還遠超解析新蛋白質結構的其他方法。
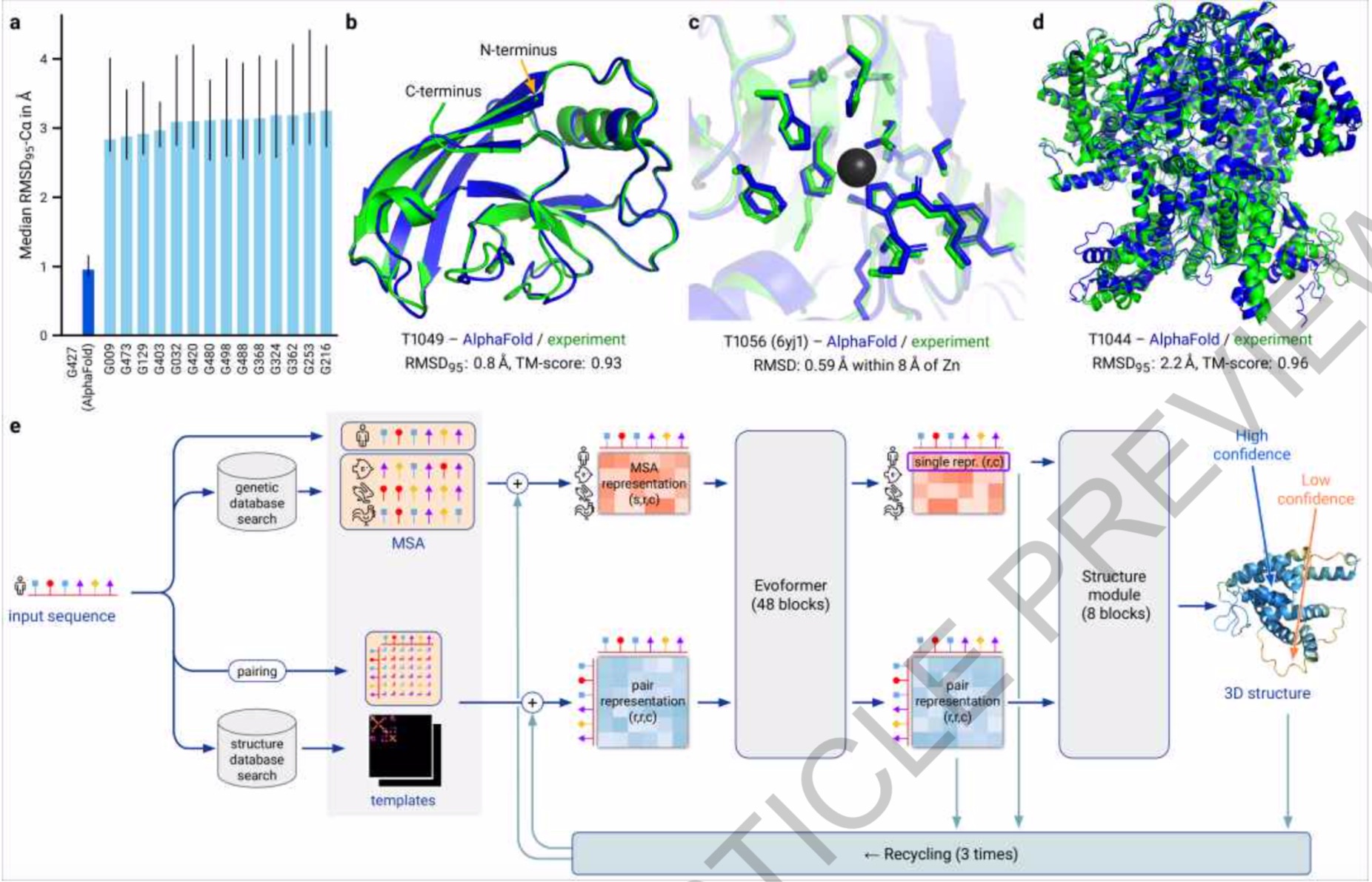
將實驗方法得到的蛋白質結構疊加在AlphaFold2的結構上,組成蛋白質主鏈骨架的疊加原子之間的距離中位數(shù)(95%的覆蓋率)為0.96埃(0.096納米)。成績排第二的方法只能達到2.8埃的準確度。
AlphaFold2的神經(jīng)網(wǎng)絡能在幾分鐘內預測出一個典型蛋白質的結構,還能預測較大蛋白質(比如一個含有2180個氨基酸、無同源結構的蛋白質)的結構。該模型能根據(jù)每個氨基酸對其預測可靠性進行精確預估,方便研究人員使用其預測結果。
AlphaFold2最終被Moult評價道,“在某種意義上,問題已經(jīng)解決了”。
值得一提的是,在最新發(fā)布的論文中,DeepMind還簡化了AlphaFold2。AlphaFold的首席研究員John Jumper說,“這個網(wǎng)絡需要幾天的計算時間來生成CASP的一些蛋白質的結構,而開源版本的速度要快16倍。根據(jù)蛋白質的大小,它可以在幾分鐘到幾小時內生成結構。”
受AlphaFold2的啟發(fā),華盛頓大學醫(yī)學院生物化學家、蛋白質設計研究所所長David Baker等人開發(fā)了RoseTTaFold。華盛頓大學醫(yī)學院官網(wǎng)對該研究的介紹稱,在高精度的蛋白質結構預測方面,Baker等人“在很大程度上重現(xiàn)了DeepMind團隊的表現(xiàn)。”
相較于AlphaFold2只解決了單個蛋白質的結構,RoseTTaFold不僅適用于簡單的蛋白質,也適用于蛋白質復合物。據(jù)介紹,RoseTTaFold利用深度學習技術,根據(jù)有限信息準確、快速地預測蛋白質結構。從結構上來看,RoseTTAFold 是一個三軌(three-track)神經(jīng)網(wǎng)絡,它可以兼顧蛋白質序列的模式、氨基酸如何相互作用以及蛋白質可能的三維結構。在這種結構中,一維、二維、三維信息來回流動,使得網(wǎng)絡能夠集中推理蛋白質的化學部分與它的折疊結構。
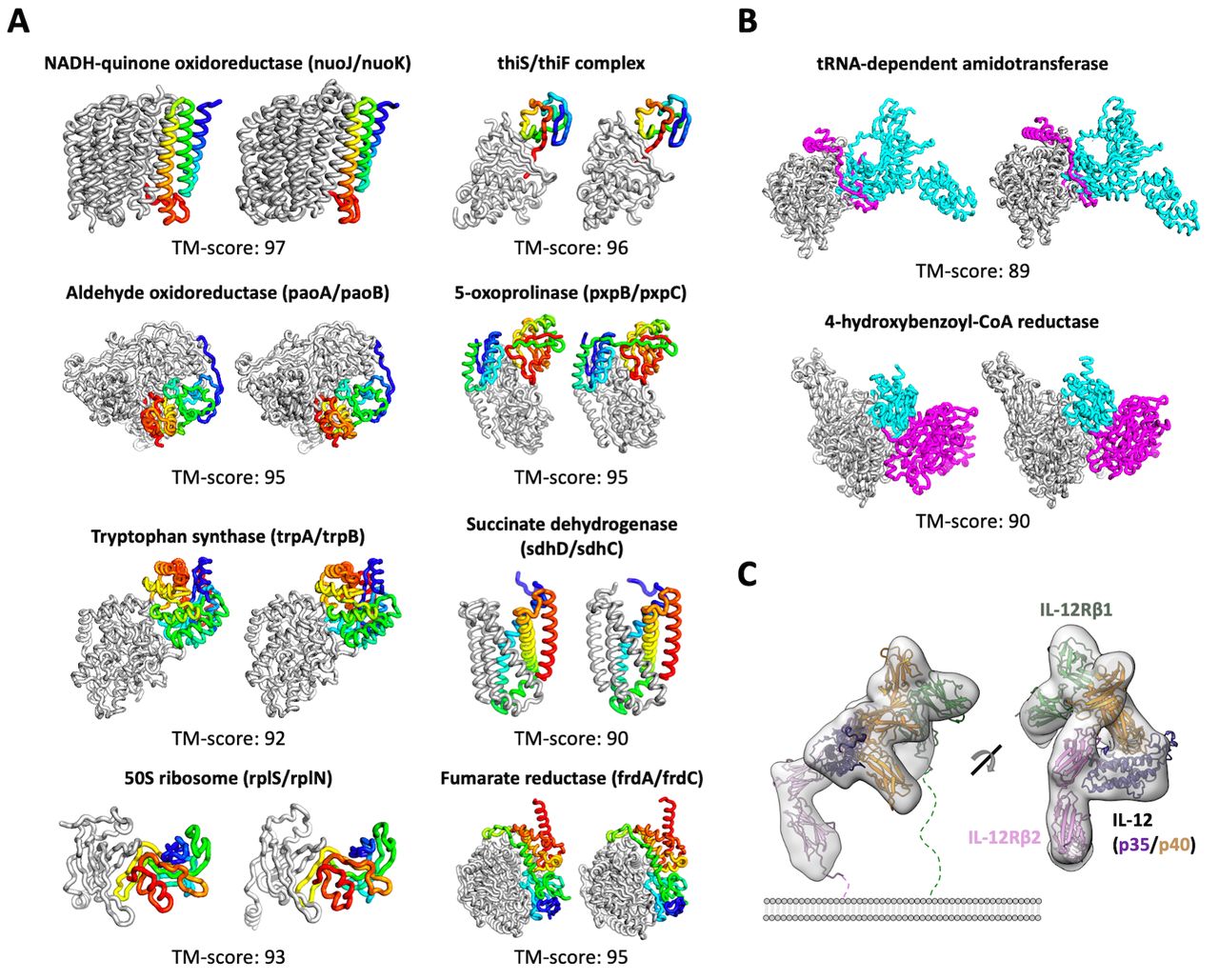
巴塞爾大學的計算結構生物學家Torsten Schwede對《科學》雜志說,許多生物功能依賴于蛋白質之間的相互作用。“直接從序列信息中處理蛋白質-蛋白質復合物的能力使其對生物醫(yī)學研究中的許多問題極具吸引力。”
Baker同時坦言,AlphaFold2的結構更加準確。但是根特大學的結構生物學家Savvas Savvides說,Bake實驗室的方法更好地捕捉到了“蛋白質結構的本質和特性”,比如識別從蛋白質側面伸出的原子串,這些特征是蛋白質之間相互作用的關鍵。
紐約大學醫(yī)學院的細胞和結構生物學家Gira Bhabha說,兩種方法都很有效。她表示,“DeepMind和Baker實驗室的進展都是驚人的,將改變我們利用蛋白質結構預測推進生物學的方式。”
開源代碼,如何促進整個科學界?
相比于去年年底帶來的震撼,這次外界更感興趣的是上述兩支團隊開源代碼這一動作。
此前的6月中旬,在Baker實驗室發(fā)布RoseTTAFold預印本三天之后,DeepMind的Hassabis在推特上表示,AlphaFold2的細節(jié)正在接受一份出版物的審查,公司將“為科學界提供廣泛的免費訪問”。
而從6月1日開始,Baker等人已經(jīng)開始挑戰(zhàn)他們的方法,讓研究人員發(fā)送來他們最令人困惑的蛋白質序列。加州大學舊金山分校的結構生物物理學家David Agard的研究小組發(fā)送了一組沒有已知類似蛋白質的氨基酸序列,幾個小時內,他的團隊就得到了一個蛋白質模型,“這可能為我們節(jié)省了一年的工作。”Agard說。
除了免費提供RoseTTaFold的代碼外,Baker團隊還建立了一個服務器,研究人員可以插入蛋白質序列并得到預測的結構。貝克說,自從上個月推出以來,該服務器已經(jīng)預測了大約500人提交的5000多種蛋白質的結構。
不過,上述兩支團隊的源代碼都是免費的,但也有觀點認為,對于沒有技術專長的研究人員來說,它可能還不是特別有用。不過,DeepMind的科學人工智能負責人Pushmeet Kohli表示,DeepMind已經(jīng)與一些選定的研究人員和組織合作,以預測特定的目標,其中包括總部位于瑞士日內瓦的非營利組織“Drugs for ignored Diseases”。“在這個領域,我們還有很多想做的事情。”
Hassabis提到,去年在CASP14大會上我們揭曉了一個可以將蛋白質3D結構預測精確到原子水平的全新AlphaFold系統(tǒng),此后我們承諾會分享我們的方法,并為科學共同體提供廣泛、免費的獲取途徑。“今天我們邁出了承諾的第一步,在《自然》期刊上分享AlphaFold的開源代碼,并發(fā)表了系統(tǒng)的完整方法論,詳盡細致說明AlphaFold是如何做到精確預測蛋白質3D結構的。作為一家致力于推動科學進步的公司,我們期待看到我們的方法將為科學界啟發(fā)出什么其他新的研究方法,也期待很快能和大家分享更多我們的新進展。”
DeepMind團隊認為,這一精準的預測算法可以讓蛋白質結構解析技術跟上基因組革命的發(fā)展步伐。
Baker團隊也提到,“我們希望這個新工具將繼續(xù)造福整個研究界。”
中國科學院合肥物質科學研究院強磁場科學中心研究員謝燦對澎湃新聞(www.6773257.com)記者表示,“總的來說,對學術界來肯定是好事,肯定會促進結構生物學和相關領域的發(fā)展。在承認學術貢獻的基礎上的開放和共享,本來就應該是學術研究最基本的要求。”
結構生物學是謝燦的“老本行”,“我當年花了8年的時間去解析一個蛋白的晶體結構,我能切身體會如果有一個精準預測蛋白結構的算法出現(xiàn),對結構生物學家意味著什么。”
但他認為,不必要擔憂這些算法的出現(xiàn)會讓結構生物學家失業(yè),在技術迭代之下,結構生物學這些年受到的沖擊太多了,“而事實上,只不過是某一個領域某一個技術在某一個歷史階段更容易出工作出成績。”謝燦認為,無論再精準的預測,終究也只是預測,“AlphaFold2不是實驗,同樣也需要實驗去證實。”
王宏偉在AlphaFold2剛出現(xiàn)之時也曾評價道,對于復雜的結構生物學問題,預測手段本身還不能號稱完全解決了問題。實驗結構生物學領域接下來需要做的一個事情是要擁抱變化,更好地與預測方法結合以及共同發(fā)展。
相關論文鏈接:
https://www.nature.com/articles/s41586-021-03819-2
https://science.sciencemag.org/content/early/2021/07/14/science.abj8754



- 報料熱線: 021-962866
- 報料郵箱: news@thepaper.cn
互聯(lián)網(wǎng)新聞信息服務許可證:31120170006
增值電信業(yè)務經(jīng)營許可證:滬B2-2017116
? 2014-2025 上海東方報業(yè)有限公司

